
Last Updated: November 9, 2025
Introduction to Sly Syndrome
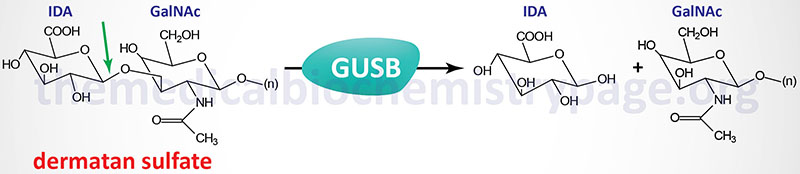
Sly syndrome is an extremely rare autosomal recessive disease that belongs to the family of disorders identified as lysosomal storage disorders, and historically as the mucopolysaccharidoses. Sly syndrome is also referred to as mucopolysaccharidosis type VII (MPS VII). Sly syndrome is characterized by the lysosomal accumulation of glucuronic acid-containing glycosaminoglycans (dermatan, heparan, and chondroitin 4- and 6-sulfates) as a consequence of deficiencies in the lysosomal hydrolase, β-glucuronidase. The worldwide frequency of Sly syndrome is estimated to be 1 in 1,000,000 births representing less than 2% of all cases of mucopolysaccharidosis.
Molecular Biology of Sly Syndrome
The β-glucuronidase gene (GUSB) resides on chromosome 7q21.11 spanning 20 kb and comprising 12 exons that generate four alternatively spliced mRNAs, each of which encode a distinct protein isoform. The longest isoform (isoform 1) derived from the GUSB gene is a 651 amino acid precursor protein. Active β-glucuronidase is a 629 amino acid protein that functions in a homotetrameric complex.
As of 2025 there were 56 pathogenic mutations characterized in the GUSB gene resulting in Sly syndrome (MPS VII). Identified mutations, which span the entirely of the GUSB gene, include missense (74% of identified mutations), nonsense, frameshift, and splice site mutations. The most commonly identified mutation in the GUSB gene worldwide is the missense mutation where Leu 176 is changed to Phe (L176F). Three other common missense mutations P408S, P415L, and A619V. A common nonsense mutation is R357Ter.
Complicating the diagnosis of the inheritance of mutant alleles of GUSB is the presence of several pseudogenes on chromosomes 5, 6, 7, 20, 22, and Y. These pseudogenes can be detected with probes from exons 2, 3, 4, 6, 7, and 11 of the wild-type GUSB gene.
Clinical Features of Sly Syndrome
Like many other lysosomal storage disorders, Sly syndrome patients exhibit a wide spectrum of clinical manifestations ranging from mild disease in adults to nonimmune hydrops fetalis (resulting in death in utero). The disease was first recognized in a patients exhibiting symptoms similar to those seen in Hurler syndrome patients (another mucopolysaccharidotic disease). Due to the spectrum of pathologies observed in Sly syndrome patients the disorder has been divided into three clinical spectrums. Group 1 disease is the neonatal non-immune hydrops fetalis (NIHF) form. Group 2 is the infantile or adolescent form with history of hydrops fetalis form. Group 3 is the infantile or adolescent without known hydrops fetalis form.
The severe neonatal form (NIHF) of Sly syndrome is the most commonly encountered form and can range from death in utero to mild or no hydrops at birth. Neonatal Sly syndrome is one of the few lysosomal storage disorders that manifests in utero or at birth.
Sly syndrome is characterized by coarse facial features, hepatosplenomegaly, cardiac valve disease, protruding sternum, and dystosis multiplex. Dystosis multiplex refers to a constellation of skeletal abnormalities and is characterized by an enlarged skull, thickened calvarium, premature closure of lamboid and sagittal sutures, shallow orbits, enlarged J-shaped sella turcica (a saddle-shaped skull structure into which sits the bottom of the pituitary gland), and abnormal spacing of the teeth with dentigerous cysts. There is anterior hypoplasia of the lumbar vertebrae, the long bone diaphyses are enlarged and an irregular appearance of the metaphyses. The epiphyseal centers not well developed, the pelvis is poorly formed with small femoral heads and coxa valga. The clavicles are short, thick and irregular and the ribs are oar shaped. Phalanges are shortened and trapezoidal in shape.
In addition to the skeletal abnormalities Sly syndrome patients also manifest with ocular features that includes corneal clouding, heavy eyebrows, visual impairment, and photosensitivity. Additional facial abnormalities include enlarged tongues, abnormal dentition, gingival hypertrophy, and widely spaced teeth. A majority of Sly syndrome patients also exhibit ear and respiratory infections. Sensorineural hearing loss has also been observed in around 40% of Sly syndrome patients.
Treatment of Sly Syndrome
Bone marrow transplantation has been attempted as a treatment for Sly syndrome with limited success. An enzyme replacement therapy (ERT) was approved by the US FDA in 2017 that involves the use of recombinant human β-glucuronidase. This drug is known as vestronidase alfa and is sold under the tradename of Mepsevii. Although administration of vestronidase alpha does improve survivability and improves quality of life it does not modify the symptoms of the disease that are present in a patient prior to the onset of treatment.