
Last Updated: October 30, 2025
Introduction to Hereditary Coproporphyria, HCP
Hereditary coproporphyria (HCP) results from deficiencies in a specific enzyme involved in the biosynthesis of heme (also called the porphyrin pathway). The term porphyria is derived from the Greek term porphura which means “purple pigment” in reference to the coloration of body fluids in patients suffering from the originally described porphyria which is now known as porphyria cutanea tarda (PCT).
The porphyrias are classified on the basis of the tissue that is the predominant site of accumulation of metabolic intermediates. These classifications are “hepatic” or “erythroid”. Each disease is also further characterized as being acute or cutaneous dependent upon the major clinical features of the disease. HCP is an autosomal dominant disorder that results from defects in oxygen-dependent coproporphyrinogen-III oxidase resulting in half-normal activity of the enzymes. HCP is classified as an acute hepatic porphyria.
Molecular Biology of Hereditary Coproporphyria
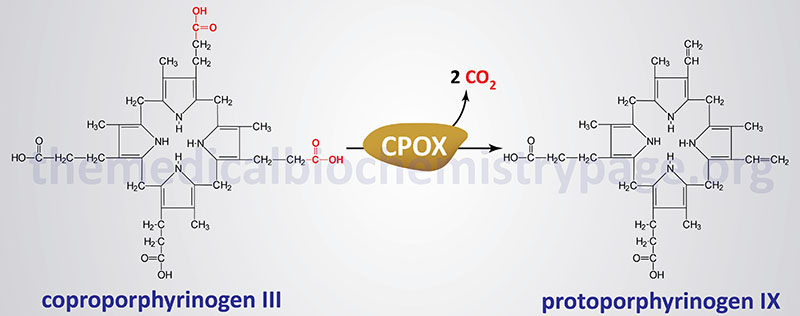
The coproporphyrinogen-III oxidase gene (CPOX) is located on chromosome 3q11.2 spanning 14 kb and encompassing 11 exons encoding a 454 amino acid precursor protein.
A variety of mutations have been identified in the CPOX gene resulting in HCP. These mutations include missense, nonsense and splice-site mutations and deletions and insertions. Most of the identified mutations are unique to a given HCP patient.
Clinical Features of HCP
The clinical manifestations of HCP are identical to those seen in patients with acute intermittent porphyria, AIP. Additionally, HCP patients experience cutaneous lesions. Many of the same factors that precipitate acute attacks in AIP patients, such as smoking, alcohol consumption, barbiturates, and steroids, result in acute attacks of porphyria in HCP patients. Symptoms of HCP remain latent until after puberty. Symptoms of HCP are more common in adult women than in men. Generally, the symptoms of HCP are milder than those seen in AIP. However, there are reported examples where HCP patients exhibit severe motor neuropathies.